
Plasma Protein Binding Rate
from tdc.benchmark_group import admet_group
from tdc.single_pred import ADME
import pandas as pd
import numpy as np
from tqdm import tqdm
from rdkit import Chem
from rdkit.ML.Descriptors.MoleculeDescriptors import MolecularDescriptorCalculator
from rdkit.Chem import AllChem
from rdkit.Chem import DataStructs
from rdkit.Chem import Descriptors
from rdkit import RDLogger
RDLogger.DisableLog('rdApp.*')
from sklearn.preprocessing import QuantileTransformer
from sklearn.ensemble import GradientBoostingRegressor
from sklearn.ensemble import GradientBoostingRegressor
from sklearn.preprocessing import QuantileTransformer
from sklearn.preprocessing import StandardScaler
from sklearn.preprocessing import RobustScaler
import sklearn.impute
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import DataStructs
from sklearn.neural_network import MLPRegressor
from tdc.single_pred import ADME
data = ADME(name = 'PPBR_AZ')
split = data.get_split()
training, test, valid = split['train'], split['test'], split['valid']
# Training, test, and validiation datasets
Found local copy...
Loading...
Done!
Feature Engineer
One set of features are the Morgan Fingerprints from RDKit, another is the chemical descriptors - the chemical physical properties.
# First, convert smiles strings to molecular information
test['mol'] = test['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
training['mol'] = training['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
valid['mol'] = valid['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
y_train, y_valid, y_test = training['Y'].to_frame(), valid['Y'].to_frame(), test['Y'].to_frame()
Chemical properties
From RDKit, there are built in molecular descriptions.
X_test_chem = [Descriptors.CalcMolDescriptors(x) for x in test['mol']]
X_train_chem = [Descriptors.CalcMolDescriptors(x) for x in training['mol']]
X_valid_chem = [Descriptors.CalcMolDescriptors(x) for x in valid['mol']]
pd.DataFrame(X_train_chem).head()
| MaxAbsEStateIndex | MaxEStateIndex | MinAbsEStateIndex | MinEStateIndex | qed | SPS | MolWt | HeavyAtomMolWt | ExactMolWt | NumValenceElectrons | ... | fr_sulfide | fr_sulfonamd | fr_sulfone | fr_term_acetylene | fr_tetrazole | fr_thiazole | fr_thiocyan | fr_thiophene | fr_unbrch_alkane | fr_urea | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 12.901867 | 12.901867 | 0.237428 | -0.861281 | 0.435219 | 11.968750 | 452.902 | 431.734 | 452.136366 | 164 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 12.059240 | 12.059240 | 0.178128 | -1.028493 | 0.687391 | 12.000000 | 307.390 | 282.190 | 307.178358 | 122 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| 2 | 12.477711 | 12.477711 | 0.063209 | -0.063209 | 0.566619 | 16.757576 | 452.515 | 424.291 | 452.217203 | 174 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 12.089598 | 12.089598 | 0.304671 | -0.558346 | 0.651154 | 14.931034 | 430.339 | 409.171 | 429.112316 | 150 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 10.217217 | 10.217217 | 0.204198 | -1.074864 | 0.716623 | 35.400000 | 347.375 | 326.207 | 347.159354 | 134 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 210 columns
There are over 200 different properties calculated by RDKit, including molecular weight, valence electrons, etc
# I'm going to want these as numpy arrays
X_test_chem = np.array(pd.DataFrame(X_test_chem))
X_train_chem = np.array(pd.DataFrame(X_train_chem))
X_valid_chem = np.array(pd.DataFrame(X_valid_chem))
# Let's see how we do without any more work
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train_chem, y_train)
GradientBoostingRegressor(random_state=1)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
GradientBoostingRegressor(random_state=1)
training_score, validation_score = regr.score(X_train_chem, y_train), regr.score(X_valid_chem, y_valid)
print(f'Training score: {training_score:.2f}, Validation score: {validation_score:.2f}')
Training score: 0.84, Validation score: 0.38
Not a bad start, but it can be better. Let’s add preprocessing to the chemical properties dataset
import matplotlib.pyplot as plt
plt.hist(y_train)
plt.show()
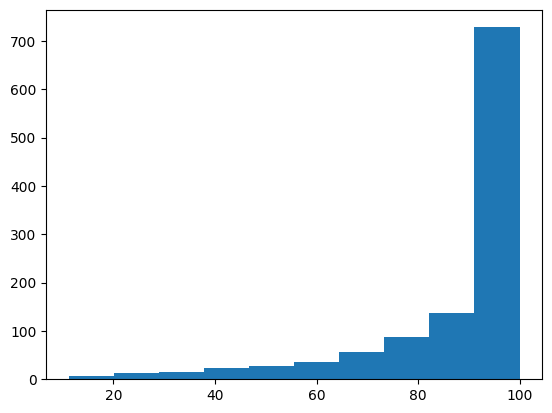
plt.hist(X_train_chem[155:160,:])
plt.show()
The target data is heavily skewed to the high values, the input data is almost always zero for a given dimension but occasionally there will be a non-zero value for some molecule.
x_scaler = QuantileTransformer(
random_state=1,
output_distribution='normal',
)
y_scaler = QuantileTransformer(
random_state=1,
output_distribution='normal',
)
X_train_scaled = x_scaler.fit_transform(X_train_chem)
X_validation_scaled = x_scaler.transform(X_valid_chem)
y_train_scaled = y_scaler.fit_transform(y_train)
y_validation_scaled = y_scaler.transform(y_valid)
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train_scaled, y_train_scaled)
training_score, validation_score = regr.score(X_train_scaled, y_train_scaled), regr.score(X_validation_scaled, y_validation_scaled)
print(f'Training score: {training_score:.2f}, Validation score: {validation_score:.2f}')
Training score: 0.81, Validation score: 0.47
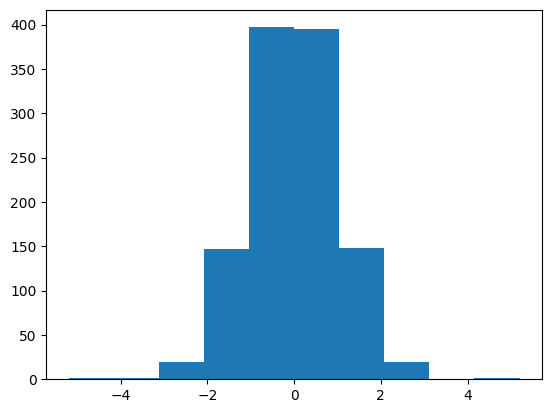
plt.hist(y_train_scaled)
plt.show()
plt.hist(X_train_scaled[155:160,:])
plt.show()
Transforming the data into a normal distribution helps train the model
Morgan fingerprinting
Another type of data to use is a more direct comparison between molecules: molecular fingerprints. These look at atoms near to one another within the molecule. These fingerprints can be used to compare molecules to one another in the dataset and judge similarity.
radius = 7 # the default radius
training['mol'] = training['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in training['mol']]
valid['mol'] = valid['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
valid_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in valid['mol']]
test['mol'] = test['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
test_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in test['mol']]
# from the fingerprints, make similarity matrices
num_molecules = len(fingerprints)
similarity_matrix = np.zeros((num_molecules, num_molecules))
num_valid_molecules = len(valid_fingerprints)
num_test_molecules= len(test_fingerprints)
num_molecules = len(fingerprints)
num_valid_molecules = len(valid_fingerprints)
num_test_molecules= len(test_fingerprints)
similarity_matrix = np.zeros((num_molecules, num_molecules))
test_similarity_matrix = np.zeros((num_test_molecules, num_molecules))
valid_similarity_matrix = np.zeros((num_valid_molecules, num_molecules))
for i in range(num_molecules):
for j in range(i, num_molecules):
similarity = DataStructs.TanimotoSimilarity(fingerprints[i], fingerprints[j])
similarity_matrix[i, j] = similarity
similarity_matrix[j, i] = similarity # Symmetric matrix
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
for i in range(num_valid_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(valid_fingerprints[i], fingerprints[j])
valid_similarity_matrix[i, j] = similarity
similarity_matrix[0:5, 0:10]
array([[1. , 0.08379888, 0.08547009, 0.09852217, 0.07537688,
0.11363636, 0.08606557, 0.10144928, 0.08 , 0.07614213],
[0.08379888, 1. , 0.05641026, 0.08024691, 0.05732484,
0.10674157, 0.05853659, 0.06508876, 0.09550562, 0.08609272],
[0.08547009, 0.05641026, 1. , 0.07798165, 0.09178744,
0.08898305, 0.1888412 , 0.08108108, 0.1038961 , 0.06666667],
[0.09852217, 0.08024691, 0.07798165, 1. , 0.07142857,
0.09178744, 0.07423581, 0.10582011, 0.12562814, 0.08426966],
[0.07537688, 0.05732484, 0.09178744, 0.07142857, 1. ,
0.04326923, 0.05803571, 0.05820106, 0.075 , 0.05142857]])
valid_similarity_matrix[0:5, 0:10]
array([[0.08982036, 0.09836066, 0.0718232 , 0.06535948, 0.06944444,
0.06936416, 0.05670103, 0.05660377, 0.08284024, 0.08571429],
[0.07352941, 0.0621118 , 0.04524887, 0.07526882, 0.06703911,
0.04225352, 0.06140351, 0.06770833, 0.05769231, 0.0441989 ],
[0.08097166, 0.07881773, 0.07307692, 0.05531915, 0.04824561,
0.07142857, 0.07407407, 0.0720339 , 0.08943089, 0.08219178],
[0.06756757, 0.05 , 0.07327586, 0.09 , 0.07731959,
0.09589041, 0.08786611, 0.05188679, 0.08675799, 0.078125 ],
[0.09876543, 0.0631068 , 0.05681818, 0.09251101, 0.05752212,
0.09311741, 0.04693141, 0.10480349, 0.08064516, 0.06756757]])
Let’s train the model on just these similarity matrices
X_train, X_valid, X_test = similarity_matrix, valid_similarity_matrix, test_similarity_matrix
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train, y_train)
training_score, validation_score = regr.score(X_train, y_train), regr.score(X_valid, y_valid)
f"Training score: {training_score:.2f}, Validation score: {validation_score:.2f}"
'Training score: 0.81, Validation score: 0.15'
It’s not better than the chemical property model, but it’s almost guaranteed to contain different information. Let’s combine them and see if the model is further improved.
# This is just an intermediate check, I'm going to skip the pre-processing.
#So I want to compare the scores with cell 25: Training score: 0.84 Validation score: 0.38
#concatenate chemical data with fingerprint data
X_train = np.concatenate((similarity_matrix, X_train_chem), axis=1)
X_valid = np.concatenate((valid_similarity_matrix, X_valid_chem), axis=1)
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train, y_train)
training_score, validation_score = regr.score(X_train, y_train), regr.score(X_valid, y_valid)
print(f'Training score: {training_score:.2f}, Validation score: {validation_score:.2f}')
Training score: 0.87, Validation score: 0.36
Determine the right radius to use
That actually made the model worse at predicting the validation dataset. I’m going to adjust how the Morgan Fingerprints are calculated to see if there’s a better way.
num_molecules = len(fingerprints)
num_valid_molecules = len(valid_fingerprints)
num_test_molecules= len(test_fingerprints)
similarity_matrix = np.zeros((num_molecules, num_molecules))
test_similarity_matrix = np.zeros((num_test_molecules, num_molecules))
valid_similarity_matrix = np.zeros((num_valid_molecules, num_molecules))
for radius in range(10):
fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in training['mol']]
#test_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in test['mol']]
valid_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in valid['mol']]
for i in range(num_molecules):
for j in range(i, num_molecules):
similarity = DataStructs.TanimotoSimilarity(fingerprints[i], fingerprints[j])
similarity_matrix[i, j] = similarity
similarity_matrix[j, i] = similarity # Symmetric matrix
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
for i in range(num_valid_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(valid_fingerprints[i], fingerprints[j])
valid_similarity_matrix[i, j] = similarity
X_train, X_valid, X_test = similarity_matrix, valid_similarity_matrix, test_similarity_matrix
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train, y_train)
print("radius: "+str(radius), regr.score(X_train, y_train), regr.score(X_valid, y_valid))
radius: 0 0.5911407088782001 -0.19957336525763902
radius: 1 0.7307129311812188 0.21432168957971698
radius: 2 0.7601903491354445 0.09446409128208133
radius: 3 0.7709970603513945 0.16536085141618106
radius: 4 0.7822654750730927 0.19376403642887308
radius: 5 0.799313228929121 0.09288487963697722
radius: 6 0.7830749173722934 0.07143858347131138
radius: 7 0.8083843165268636 0.15329793225288801
radius: 8 0.7991026185815263 0.10111769580646157
radius: 9 0.7923410240659565 -0.003452249174486388
Looks like a radius of 1 is the right range for this dataset. So we’re looking at the central atom and it’s most immediate neighbors.
Put it all together, train the model
I want to bring the chemical descriptions, the molecular fingerprints, and the preprocessing together
## Get the data
training, test, valid = split['train'], split['test'], split['valid']
test['mol'] = test['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
training['mol'] = training['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
valid['mol'] = valid['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
y_train, y_valid, y_test = training['Y'].to_frame(), valid['Y'].to_frame(), test['Y'].to_frame()
## Add Chemical properties
X_test_chem = np.array(pd.DataFrame([Descriptors.CalcMolDescriptors(x) for x in test['mol']]))
X_train_chem =np.array(pd.DataFrame([Descriptors.CalcMolDescriptors(x) for x in training['mol']]))
X_valid_chem =np.array(pd.DataFrame([Descriptors.CalcMolDescriptors(x) for x in valid['mol']]))
## Add Morgan fingerprints
radius = 1
fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in training['mol']]
test_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in test['mol']]
valid_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in valid['mol']]
num_molecules = len(fingerprints)
num_valid_molecules = len(valid_fingerprints)
num_test_molecules= len(test_fingerprints)
similarity_matrix = np.zeros((num_molecules, num_molecules))
test_similarity_matrix = np.zeros((num_test_molecules, num_molecules))
valid_similarity_matrix = np.zeros((num_valid_molecules, num_molecules))
for i in range(num_molecules):
for j in range(i, num_molecules):
similarity = DataStructs.TanimotoSimilarity(fingerprints[i], fingerprints[j])
similarity_matrix[i, j] = similarity
similarity_matrix[j, i] = similarity # Symmetric matrix
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
for i in range(num_valid_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(valid_fingerprints[i], fingerprints[j])
valid_similarity_matrix[i, j] = similarity
X_train = np.concatenate((similarity_matrix, X_train_chem), axis=1)
X_valid = np.concatenate((valid_similarity_matrix, X_valid_chem), axis=1)
X_test = np.concatenate((test_similarity_matrix, X_test_chem), axis=1)
### Preprocessing
x_scaler = QuantileTransformer(
random_state=1,
output_distribution='normal',
)
y_scaler = QuantileTransformer(
random_state=1,
output_distribution='normal',
)
X_train_scaled = x_scaler.fit_transform(X_train) #Dont' cheat, fit the transform on the training data
X_validation_scaled = x_scaler.transform(X_valid)
y_train_scaled = y_scaler.fit_transform(y_train) #Dont' cheat, fit the transform on the training data
y_validation_scaled = y_scaler.transform(y_valid)
### Train and evaulate the model
regr = GradientBoostingRegressor(
random_state=1,
)
regr.fit(X_train_scaled, y_train_scaled)
training_score, validation_score = regr.score(X_train_scaled, y_train_scaled), regr.score(X_validation_scaled, y_validation_scaled)
print(f'Training score: {training_score:.2f}, Validation score: {validation_score:.2f}')
Training score: 0.83, Validation score: 0.44
Comapred to where we started (validation score = 0.38) this is a moderate improvement. Let’s submit!
Using the submission framework
Copying submission method from MapLight. Thanks Jim!
benchmark_config = {
'ppbr_az': ('regression', False),
}
group = admet_group(path = 'data/')
for admet_benchmark in [list(benchmark_config.keys())[0]]:
predictions_list = []
for seed in tqdm([1, 2, 3, 4, 5]):
benchmark = group.get(admet_benchmark)
predictions = {}
name = benchmark['name']
training, test = benchmark['train_val'], benchmark['test']
test['mol'] = test['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
training['mol'] = training['Drug'].apply(lambda x: Chem.MolFromSmiles(x))
radius=1
fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in training['mol']]
test_fingerprints = [AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=2048) for mol in test['mol']]
num_molecules = len(fingerprints)
num_test_molecules = len(test_fingerprints)
similarity_matrix = np.zeros((num_molecules, num_molecules))
test_similarity_matrix = np.zeros((num_test_molecules, num_molecules))
for i in range(num_molecules):
for j in range(i, num_molecules):
similarity = DataStructs.TanimotoSimilarity(fingerprints[i], fingerprints[j])
similarity_matrix[i, j] = similarity
similarity_matrix[j, i] = similarity # Symmetric matrix
for i in range(num_test_molecules):
for j in range(0, num_molecules):
similarity = DataStructs.TanimotoSimilarity(test_fingerprints[i], fingerprints[j])
test_similarity_matrix[i, j] = similarity
X_train, X_test = similarity_matrix, test_similarity_matrix
X_train_chem = np.array(pd.DataFrame([Descriptors.CalcMolDescriptors(x) for x in training['mol']]))
X_test_chem = np.array(pd.DataFrame([Descriptors.CalcMolDescriptors(x) for x in test['mol']]))
X_train = np.concatenate((X_train, X_train_chem), axis=1)
X_test = np.concatenate((X_test, X_test_chem), axis=1)
qt = QuantileTransformer(
random_state=seed,
output_distribution='normal',
)
y_train, y_test = training['Y'].to_frame(), test['Y'].to_frame()
y_train = qt.fit_transform(y_train)
y_test = qt.transform(y_test)
scaler = QuantileTransformer(
random_state=seed,
output_distribution='normal',
)
X_train = scaler.fit_transform(X_train)
X_test = scaler.transform(X_test)
regr = GradientBoostingRegressor(
random_state=seed,
)
regr.fit(X_train, y_train)
y_pred = regr.predict(X_test).reshape(-1,1)
y_pred_test = qt.inverse_transform(y_pred).reshape(-1)
# --------------------------------------------- #
prediction_dict = {name: y_pred_test}
predictions_list.append(prediction_dict)
results = group.evaluate_many(predictions_list)
print('\n\n{}'.format(results))
Found local copy...
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████| 5/5 [10:37<00:00, 127.51s/it]
{'ppbr_az': [7.441, 0.024]}
As of 2024/07/09, this is a first place score on the leaderboard!
| The current leader is MapLight + GNN, Jim Notwell, 7.526 ± 0.106 |